


SÍNDROME DE GOOD – RELATO DE CASO
Resumo
A Síndrome de Good, associação entre timoma e imunodeficiência, cursa com infecções recorrentes e na avaliação imunológica, redução ou ausência de linfócitos B, hipogamaglobulinemia e defeitos na imunidade celular. Descrevemos uma paciente de 58 anos com sinusites e pneumonias recorrentes e antecedente de estrongiloidíase disseminada. Tomografia de tórax evidenciou massa mediastinal ocupando loja tímica e posteriormente bronquiectasias em lobos superiores. Realizada exérese e histopatológico da massa confirmando timoma. Na avaliação imunológica: hipogamaglobulinemia e redução de linfócitos B. Avaliar linfócitos T e B e quantificar imunoglobulinas deve ser rotina nos pacientes com timoma, permitindo identificar precocemente os portadores da síndrome.
Descritores (Palavras-chave)
Timoma; Agamaglobulinemia; Síndromes de Imunodeficiência; Infecção
Introdução
A associação entre timoma e imunodeficiência foi primeiro descrita por Robert Good em 1954, sendo então denominada Síndrome de Good.1 É uma condição rara que acomete mais adultos com idade entre 40 e 70 anos. Caracteriza-se pela presença de timoma e quadros infecciosos de repetição, onde, à avaliação imunológica, verifica-se redução ou ausência de células B no sangue periférico, hipogamaglobulinemia e defeitos na imunidade mediada por células.2 A síndrome ocorre em 7 a 13% dos pacientes com hipogamaglobulinemia de início na idade adulta.3
Em pacientes com timoma, ,a incidência de hipogamaglobulinemia é de 6-11%.4 O Quadro 1 descreve características da síndrome – manifestações clínicas e complicações, achados laboratoriais e opções de tratamento.
Quadro 1– Características importantes da Síndrome de Good
| DEFINIÇÃO | IMUNODEFICIÊNCIA COM TIMOMA |
| Manifestações clínicas e complicações | Susceptibilidade aumentada a infecções bacterianas, especialmente organismos encapsulados e oportunistas (vírus e fungos); doenças auto-imunes, como miastenia gravis, neutropenia, diabetes mellitus, polimiosite, aplasia pura de células vermelhas e anemia. |
| Achados laboratoriais | Hipogamaglobulinemia, redução ou ausência de células B, linfopenia TCD4+, redução de resposta proliferativa de células T frente a mitógenos. |
| Opções de tratamento | Ressecção do timoma se maligno, invasivo ou obstrutivo; reposição das imunoglobulinas; antibiótico quando indicado; agentes imunossupressores se presença de doença auto-imune. |
Adaptado de AGARWAL; CUNNINGHAM-RUNDLES, 2007.2
O presente relato descreve caso clínico de paciente com síndrome de Good, acompanhado por revisão de literatura.
Relato do caso
Paciente do sexo feminino iniciou quadro de infecções respiratórias de repetição (sinusites e pneumonias) aos 55 anos. No mesmo ano ficou internada durante sete dias, devido à estrongiloidíase disseminada. À avaliação radiológica, foi observada lesão expansiva com densidade de partes moles em mediastino anterior, ocupando loja tímica (Figura 1). Foi submetida à cirurgia, para retirada da massa, e radioterapia. O exame histopatológico confirmou tratar-se de timoma.
Apresentava antecedente de eosinofilia desde a infância, tendo recebido vários tratamentos anti-parasitários, especialmente pelo achado persistente, ao exame de fezes, do parasita Strongiloides stercoralis e do protozoário Blastocystis hominis. Relatava, também, quadro de sibilância recorrente, fazendo uso de corticosteroide inalatório associado a beta2-agonista de ação prolongada.
Dois anos após a timectomia, foi encaminhada para avaliação imunológica. Nesta ocasião já apresentava bronquiectasias em lobos superiores de pulmão direito e esquerdo. A investigação imunológica evidenciou quadro de hipogamaglobulinemia e redução da subpopulação de linfócitos B (Quadro 2), sendo iniciado reposição de gamaglobulina intravenosa mensalmente.
Quadro 2 – Exames laboratoriais
| Hemograma
Série vermelha |
Hb 11,1 g/dl Htc 33,8% VCM 87,9fl | |
| Leucograma | Leuc 14.100 cel/mm3 (1% bast; 57% seg; 22% eos; 14% linf; 6% mono) | |
| Plaquetas | 806.000/mm3 | |
| Subpopulação de linfócitos | Valores do paciente
% – contagem absoluta linfócitos/mm3 |
Valores de referência
Contagem absoluta linfócitos/mm3 |
| CD3 | 78% – 1540/ mm3 | 844-1943 |
| CD4 | 35,3% – 697/ mm3 | 476-1136 |
| CD8 | 38,8% – 766/ mm3 | 248-724 |
| CD4/CD8 | 0,9 | 0,9-2,6 |
| CD19 | 0,2% – 4/ mm3 | 138-544 |
| Imunoglobulinas | ||
| IgG | 169 mg/dl | 739-1390 mg/dl |
| IgA | 46 mg/dl | 84-354 mg/dl |
| IgM | 10 mg/dl | 81-167 mg/dl |
| IgE | 3 UI/ml | |
Discussão
A associação entre timoma e hipogamaglobulinemia era definida como um subtipo de imunodeficiência comum variável (ICV), entretanto, o reduzido número de células B periféricas, notado na síndrome de Good, fez com que a mesma fosse classificada como uma imunodeficiência distinta, uma vez que o que se observa na ICV é um impedimento na maturação de células B e não redução tão acentuada das mesmas como visto nesta síndrome.5,6,7,8
É uma condição rara, com poucos casos reportados na literatura. KELESIDIS &YANG (2010), em revisão sistemática, descreveram achados clínicos, laboratoriais e imunológicos de 152 pacientes, sendo que 47% eram europeus.8 O início dos sintomas é tardio, como se verifica no presente relato, sendo a média de idade dos pacientes ao início dos sintomas de 56 anos (29-75a) e a idade ao diagnóstico de 62 anos (41-79a), só havendo um relato na faixa etária pediátrica.9,10,11,12 Não há diferença significativa de acometimento entre sexos.
Os timomas são neoplasias de crescimento lento que correspondem a 20 a 30 % das massas mediastinais em adultos, aparecendo na radiografia de tórax como uma massa lobulada bem definida.12 Podem ser, no entanto, não visualizados ao exame radiográfico em aproximadamente 20-24% dos casos e a tomografia de tórax pode ser mais sensível.13 Tipicamente são um achado incidental, assintomático, porém um terço dos pacientes podem manifestar sintomas de dor torácica, tosse ou dispnéia relacionados à compressão pelo tumor ou invasão.12 A maioria dos timomas ressecados na síndrome de Good são benignos, encapsulados e 75% do tipo fusiforme. Metástase é incomum, mas são comumente associados a doenças sistêmicas e autoimunes como aplasia pura da série vermelha, hipogamaglobulinemia, pancitopenia, doenças do colágeno e miastenia gravis.8,13,14,15 A ressecção do timoma não reverte as anormalidades imunológicas, sugerindo, por exemplo, que a hipogamaglobulinemia não é diretamente causada pelo timoma, mas secundária a processos autoimunes ou imunoregulatórios, como será discutido adiante. A imunodeficiência pode preceder ou ocorrer após diagnóstico do timoma. Há casos descritos de sintomas infecciosos e hipogamaglobulinemia com início até 18 anos após a timectomia.16 No caso descrito, a paciente iniciou os quadros infecciosos de repetição antes da detecção e ressecção da massa mediastinal, o que sugere um possível comprometimento imunológico prévio.
Em relação aos principais achados imunológicos na Síndrome de Good, encontramos hipogamaglobulinemia e redução ou ausência de células B, podendo haver anormalidade na relação entre CD4/CD8, linfopenia das células T CD4 e redução da resposta da célula T frente aos mitógenos.3,17 A paciente relatada apresentava níveis extremamente reduzidos de células B maduras, o que foi descrito em 87% dos casos reportados na literatura, sendo citada, também, ausência de células pré-B em amostras de medula óssea de alguns pacientes com esta síndrome.3,17 Em relação às anormalidades na contagem e função das células T, os dados disponíveis nas publicações são mais limitados e, embora sejam relatados em um grande número de pacientes, o percentual de células T e a relação CD4/CD8 foram normais na paciente estudada. A resposta linfoproliferativa dos linfócitos, frente a mitógenos, não foi avaliada.
A patogênese desta associação, timoma e deficiência de anticorpos, é desconhecida. Há, entretanto, hipóteses que sugerem aprisionamento de células pré-B, impedimento na maturação de precursores eritróides e mielóides, distúrbios na diferenciação de células B devido a fatores humorais derivados da medula óssea e disfunção de células T que causam disfunção na diferenciação da linhagem de célula B.6,18
As infecções que os pacientes apresentam são resultado dos defeitos na imunidade humoral e celular. TARR et al., revisando os quadros infecciosos e microorganismos isolados em 51 casos de Síndrome de Good, descreveu as infecções recorrentes do trato respiratório superior e inferior como as mais frequentes.10 As bactérias encapsuladas foram os patógenos mais isolados, Haemophilus influenzae em 24% dos casos e Streptococcus pneumoniae em 8%. Bronquiectasia esteve presente em 7 casos nesta série, com isolamento de Pseudomonas spp, Klebsiella spp e outros Gram negativos. Giardia lamblia e bactérias patogênicas entéricas (Salmonella spp, Campylobacter jejeuni) foram achadas em pacientes com diarréia crônica. Candidíase mucocutânea crônica foi diagnosticada em 24% dos casos. As infecções virais estiveram presentes em 40% dos pacientes, sendo o citomegalovírus o patógeno mais comum (24% dos casos).7 A paciente deste relato apresentava quadros infecciosos em via aérea, provavelmente por germes encapsulados, que são os mais comumente implicados neste tipo de infecção, porém não houve identificação de agente infeccioso nos episódios.
A infestação intestinal persistente encontrada nesta paciente, por Strongyloides stercoralis, não foi verificada em outros relatos de síndrome de Good, com exceção de um artigo nacional que relatou associação entre timoma e estrongiloidíase grave.19 O paciente avaliado era do sexo masculino, apresentava 59 anos e foi a óbito 6 dias após admissão devido a quadro pulmonar infeccioso grave, acompanhado por numerosos episódios de diarréia e anemia grave. No laudo de necropsia observou-se a presença do timoma encapsulado e, no intestino, intenso infiltrado inflamatório, com presença de larvas rabditóides e ovos de estrongilóides nas criptas. Pela rápida evolução, não se comprovou hipogamaglobulinemia e não havia descrição do número total de linfócitos, porém constataram baixos níveis de globulinas à eletroforese de proteínas. A associação de estrongiloidíase e outros defeitos de anticorpos, como ICV e deficiência de IgA, também são descritos como casos raros, uma vez que estados de hiperinfecção com risco de disseminação deste parasita estão mais associados com impedimento da imunidade celular e em pacientes em uso de corticoterapia sistêmica.10
O tratamento do timoma inclui a ressecção do timo para prevenir crescimento local invasivo e metástases, além de radioterapia e quimioterapia combinada se doença tumoral avançada em estágio 3 ou 4.16 A deficiência de anticorpos requer reposição de gamaglobulina. Como em outros casos de hipogamaglobulinemia, a reposição de gamaglobulina controla melhor a incidência das infecções, com redução das hospitalizações e do uso de antibióticos.17 O prognóstico, no entanto, parece ser menos favorável quando se compara a pacientes portadores de agamaglobulinemia ligada ao X e ICV.9,18 As principais causas de morte são resultado de infecção, doença auto-imune ou complicações hematológicas.3
A associação entre timoma e imunodeficiência é rara, mas os diversos relatos vêm contribuindo para um melhor entendimento desta doença. A investigação imunológica, incluindo avaliação das subpopulações de células T e B e quantificação das imunoglobulinas, deve ser considerada na avaliação diagnóstica de rotina de pacientes com timoma, de forma a propiciar reconhecimento precoce dos portadores da síndrome de Good.
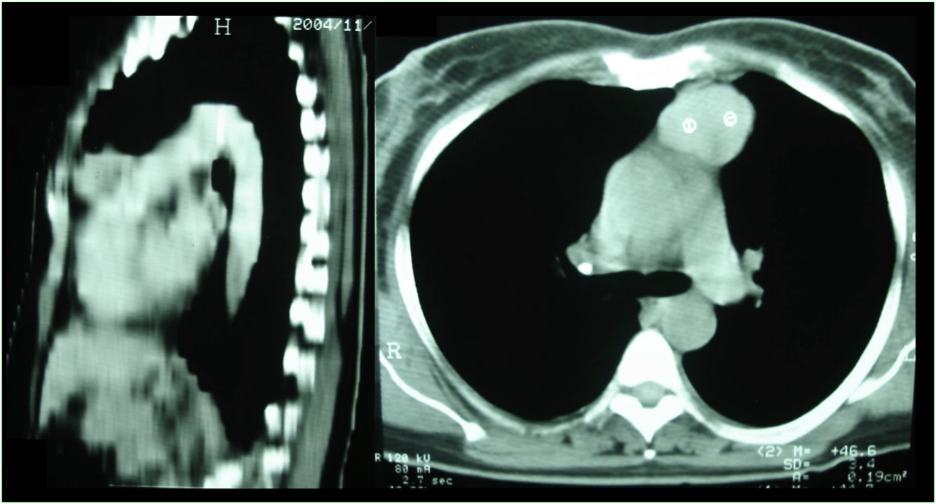
Figura 1 Tomografia computadorizada de tórax evidenciando massa em mediastino anterior à esquerda, ocupando topografia de loja tímica e medindo cerca de 5,0 x 4,0 cm nos seus maiores eixos.
Referências
- Good RA. Agammaglobulinemia: a provocative experiment of nature. Bull Univ Minn. 1954;26:1-19.
- Agarwal S, Cunningham-Rundles C. Thymoma and immunodeficiency (Good syndrome): a report of 2 unusual cases and review of the literature. Ann Allergy Asthma Immunol. 2007;98(2):185-90.
- Kelleher P, Misbah SA. What is Good’s syndrome? Immunological abnormalities in patients with thymoma. J Clin Pathol. 2003;56(1):12-6.
- Rosenow EC, Hurley BT. Disorders of the thymus. A review. Arch Intern Med. 1984;144(4):763-70.
- Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(5 Suppl 1):S1-63.
- Joven M, Palalay MP, Sonido C. Case report and literature review on Good’s syndrome, a form of acquired immunodeficiency associated with thymomas. Hawaii J Med Public Health. 2013;72 56–62.
- Notarangelo L, Casanova JL, Conley ME, Chapel H, Fischer A, Puck J et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Meeting in Budapest, 2005. J Allergy Clin Immunol. 2006;117(4):883-96.
- Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
- Kelesidis T, Yang O. Good’s syndrome remains a mystery after 55 years: a systematic review of the scientific evidence. Clin Immunol. 2010;135:347-63.
- Tarr PE, Sneller MC, Mechanic LJ, Economides A, Eger CM, Strober W et al. Infections in patients with immunodeficiency with thymoma (Good syndrome). Report of 5 cases and review of the literature. Medicine (Baltimore). 2001;80(2):123-33.
- Sicherer SH, Cabana MD, Perlman EJ, Lederman HM, Matsakis RR, Winkelstein JA. Thymoma and cellular immune deficiency in an adolescent. Pediatr Allergy Immunol. 1998;9(1):49-52.
- Duwe BV, Sterman DH, Musani AI. Tumors of the mediastinum. Chest. 2005;128(4):2893-909.
- Arend SM, Dik H, van Dissel JT. Good’s syndrome: the association of thymoma and hypogammaglobulinemia. Clin Infect Dis. 2001;32(2):323-5.
- Thompson C, Steensma D. Pure red cell aplasia associated with thymoma: clinical insights from a 50-year single institution experience. Br J Haematol. 2006;135:405–7.
- Frith J, Toller-Artis E, Tcheurekdjian H, Hostoffer R. Good syndrome and polymyositis. Ann Allergy Asthma Immunol. 2014;112(5):478.
- Raschal S, Siegel JN, Huml J, Richmond GW. Hypogammaglobulinemia and anemia 18 years after thymoma resection. J Allergy Clin Immunol. 1997;100(6 Pt 1):846-8.
- Fijolek J, Wiatr E, Demkow U, Orlowsk TM. Immunological disturbances in Good’s syndrome. Clin Invest Med. 2009;32:301–6.
- Oritani K, Kincade PW, Tomiyama Y. Limitin: an interferon-like cytokine without myeloerythroid suppressive properties. J Mol Med. 2001;79(4):168-74.
- Godoy P, Campos CMC, Costa G. Associação timoma e estrongiloidíase intestinal grave. Rev Soc Bras Med Trop. 1998;131: 481-5.
Autores
Faradiba Sarquis Serpa: Mestre em Imunologia Clínica pela Universidade Federal do Rio Janeiro – UFRJ. – (Professora Assistente de Clínica Médica da Escola Superior de Ciências da Santa Casa de Misericórdia de Vitória – ES).
Joseane Chiabai: Mestre em Ciências pela Universidade de São Paulo – USP. – (Professora Auxiliar de Pediatria da Universidade Federal do Espírito Santo – UFES).
Marcos Daniel de Deus Filho: Mestre – (Professor Assistente de Clínica Médica da Universidade Federal do Espírito Santo – UFES).
Firmino Braga Neto: Especialista em Pneumologia. – (Professor de Clínica Médica da Escola Superior de Ciências da Santa Casa de Misericórdia de Vitória-ES).
